Cómo lo desentrañamos
Cada ADN cuenta una historia
Cómo lo desentrañamos
En Genera, creemos que conocer tu historia es transformador, y entender tu linaje genético es una de las mejores maneras de hacerlo. Para ayudarte en este viaje, desarrollamos este material explicativo para que, además de descubrir más sobre su ADN, pueda entender cómo funcionan nuestros análisis y metodologías.
Los análisis de Genera se realizan en nuestro parque tecnológico, el mayor de América Latina. Los resultados se obtienen a partir del análisis de hasta 700.000 regiones de ADN: los marcadores genéticos seleccionados se comparan con nuestra base de datos, que contiene más de 9.000 muestras de referencia, obtenidas a través de decenas de estudios científicos y de 300 poblaciones de todo el mundo.
Parte I
¿Qué es lo que buscamos?
El ADN (sigla en inglés para “ácido desoxirribonucleico”) es la molécula que se encuentra en el interior de prácticamente todas las células que componen nuestro cuerpo y que contiene toda la información genética que nos compone y gestiona a cada uno de nosotros. Es esta información la que define y regula cómo somos, ya sea en términos de características físicas como la altura, el color de los ojos y del pelo, o en términos de rasgos de personalidad y predisposición a diferentes enfermedades o en el control del metabolismo y el funcionamiento de los órganos. Cada ser humano recibe la mitad de su ADN del de su madre y la otra mitad de su padre, de tal manera que, a lo largo de las generaciones, cada uno de nosotros conserva información sobre los que nos precedieron.

Desde el origen del hombre moderno en África, muchas cosas han cambiado en la dinámica de las poblaciones. Los desplazamientos no eran tan fáciles como ahora, por lo que en el pasado las poblaciones solían permanecer aisladas y con poco flujo genético (migración de un individuo de una población a otra). Este hecho, sumado a la evolución, hizo que las poblaciones antiguas se diferenciaran genéticamente entre sí, ya que las variaciones genéticas quedaban confinadas a un espacio geográfico determinado, siendo heredadas sólo por los individuos de una misma región. Esta dinámica se ha mantenido intacta durante mucho tiempo, y algunas poblaciones actuales siguen teniendo un perfil genético muy característico, especialmente las situadas en regiones geográficamente aisladas.
Existen regiones del ADN que se han alterado más que otras a lo largo de las generaciones y suelen utilizarse como marcadores genéticos. En el caso de los test de ancestralidad, analizamos los SNP (polimorfismo de un solo nucleótido). Estos marcadores específicos consisten en mutaciones de un solo nucleótido, es decir, en uno de los cuatro tipos de moléculas que componen el ADN, representadas por las letras A, T, C y G. Por lo tanto, un SNP es una variación de un par de letras en la secuencia del gen.
Por ejemplo, algunas personas pueden tener una secuencia ATTC, mientras que otras tienen AGTC. Este intercambio de la letra T por la G es un SNP. Como tienen una tasa de mutación muy baja, el intercambio de letras se considera un acontecimiento raro y tarda mucho tiempo en conseguir una frecuencia considerable en las poblaciones. El conjunto de frecuencias poblacionales de miles de SNPs nos da un perfil genético para cada población.

Parte II
Nuestra base de datos
Para comprender la composición estimada de su ascendencia, comparamos un conjunto de SNP informativos de su ADN con perfiles de población de 44 regiones diferentes. Varias de estas regiones se subdividen cualitativamente, indicando la mayor probabilidad de origen del ADN. Para calcular esta probabilidad, trabajamos con una base de datos genéticos de más de 9.000 individuos que representan a 300 poblaciones de todo el mundo, cuyos genotipos se recuperaron de publicaciones científicas y bases de datos públicas. A partir de estos análisis, los datos se agruparon en 107 regiones y subregiones, teniendo en cuenta la similitud genética y la historia de las poblaciones.
En muchos casos, una región geográfica se compone de poblaciones diferentes entre sí, pero similares a las de otros lugares del planeta, lo que refleja las migraciones, invasiones, diásporas y colonizaciones que han tenido lugar a lo largo de la historia. Es importante que estas regiones utilizadas en la base sean lo suficientemente diferentes entre sí para que la comparación sea válida.
El siguiente gráfico muestra el perfil genético de siete grandes grupos de población (África, América, Asia, Europa, Diáspora Judía, Oceanía y Oriente Medio), de los cuales hay 107 regiones y subregiones. Dado que las poblaciones están muy alejadas geográficamente, los perfiles acaban siendo muy distintos entre sí, lo que se representa con colores casi homogéneos en cada bloque.
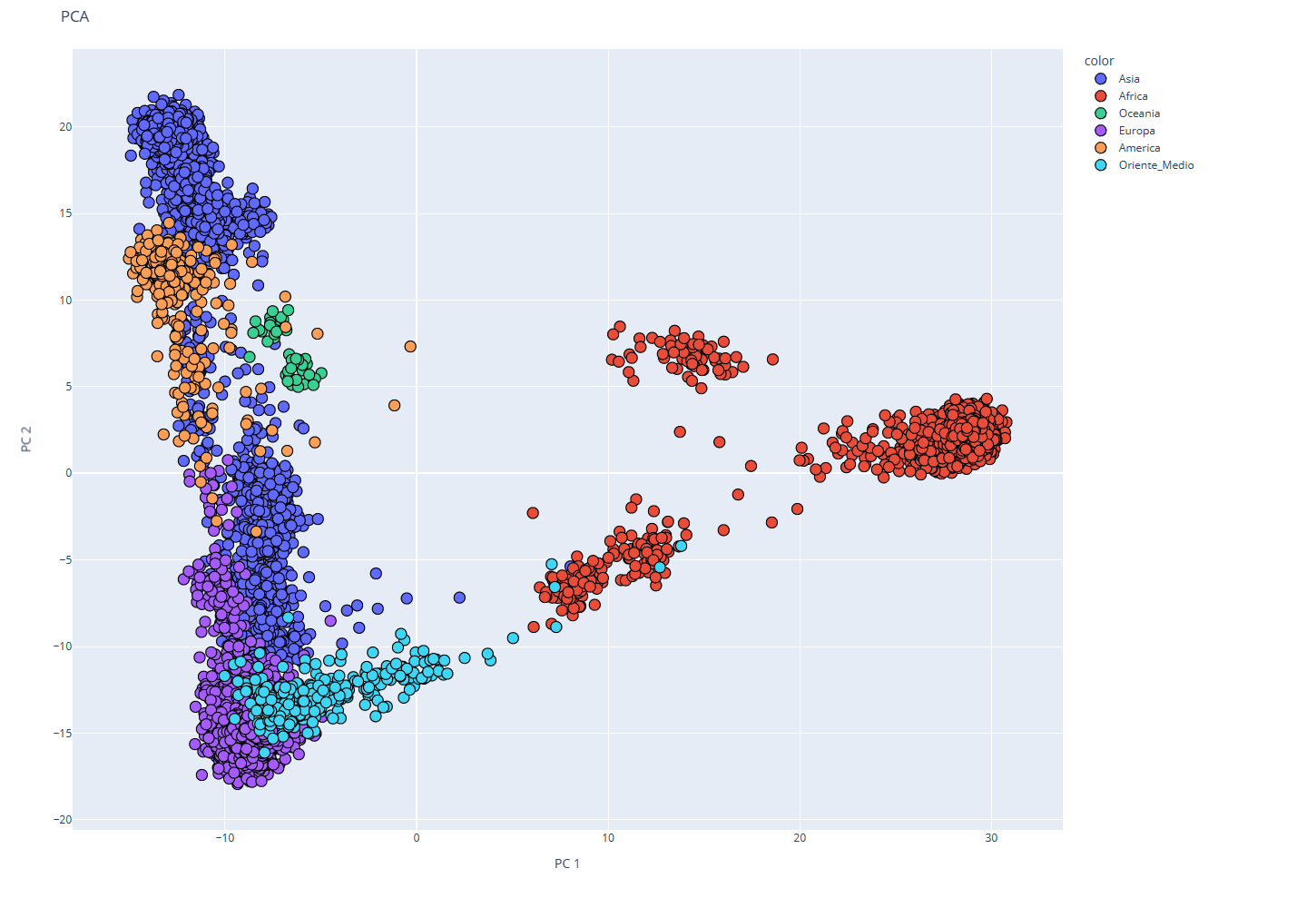
A continuación, se describen las regiones y subregiones que componen cada grupo en nuestro test:
África
- Cuerno de África
- Norte de Etiópia
- Centro-Sur de Etiopía
- Somalia
- África Oriental
- Kenia Occidental
- Región de los Grandes Lagos (pueblos bantúes del este)
- Nilotas
- Sudán del Sur y suroeste de Etiopía
- Etiopía occidental
- Sudán
- Khoisan
- África Occidental
- Senegambia
- Mandé
- Costa de Marfil
- Bayaka
- Mbuti
- Madagascar
Europa
- Italia
- Norte da Italia
- Centro-Sur de Italia
- Iberia
- Cáucaso
- Armenia
- Georgia
- Cáucaso Norte (Rusia)
- Anatolia
- Balcanes
- Bulgaria y Macedonia del Norte
- Croacia y Bosnia-Herzegovina
- Grecia
- Rumanía y Moldavia
- Serbia y Montenegro
- Laponia y Idel-Ural
- Rusia (región de Idel-Ural)
- Laponia (pueblos sami)
- Europa del Este
- Hungría
- Lituania, Letonia y Estonia
- Rusia centro-occidental
- Polonia y Eslovaquia
- Ucrania y Bielorrusia
- Europa del Este
- Islas Británicas
- Alemania, Francia, Países Bajos
- Islas Vascas
- Cerdeña
- Fenoscandia
- Ashkenazim
- Sefaradim
Asia
- Asia Central
- Turkmenistán, Uzbekistán, Kazajistán y Kirguistán
- Tayikistán
- Pakistán (provincia de Baluchistán)
- Asia del Sur
- Bangladesh
- India y Sri Lanka
- Pakistán (Pueblos Sindhi, Burusho y Pathan)
- Sudeste de Asia
- Laos
- Malasia
- Myanmar
- Vietnam
- China Dai
- Taiwán
- Islas Sonda
- Siberia
- Siberia del Norte
- Siberia occidental
- Siberia oriental y Mongolia
- Japón y Corea
- Japón
- Corea
- Filipinas y Brunéi
- Filipinas
- Brunéi
- Isla de Luzón (Pueblos Kankanaey)
- China Han
- Mongolia
- Tíbet
Américas
- América del Norte
- Centroamérica
- América Andina
- Lago Titicaca (Uros, Quechua y Aimara)
- Región Andina Central (Pueblos Aimaras y Quechua)
- Amazonas
- Tupi
- Tupi Ariquém
- Tupi Mondé
- Patagonia
Oriente Medio
- Levante
- Jordania
- Siria y Líbano
- Arabia y Egipto
- Egipto
- Israel
- Arabia Saudí, Yemen y EAU
- Magreb
- Argelia y Túnez
- Marruecos
- Mizrahim
- Teimanim
Oceanía
- Melanesia
- Islas Salomón
- Papúa Nueva Guinea
–
Al definir las poblaciones y las regiones, también se tuvo en cuenta la historia particular de la composición alélica de la población americana. Esto significa que hemos tenido especial cuidado a la hora de definir las poblaciones amerindias, de modo que hemos podido dividirlas en seis grupos y cuatro subgrupos: Tupi (Tupi Mondé y Tupi Ariquém), Amazonas, América Andina (Lago Titicaca y Región Central de los Andes), Patagonia, América Central y América del Norte).
Las subregiones de cada lugar presentan un resultado cualitativo de la ancestralidad de una persona, diferente de las regiones, que presentan valores cuantitativos. El resultado cualitativo sirve para informar, dentro de cada región, de cuál es el lugar de origen más probable del ADN detectado.


¿Cómo se deduce la ancestralidad de una persona?
A partir de las frecuencias alélicas de cada SNP para cada una de las poblaciones, calculamos, por el método de máxima verosimilitud, la composición ancestral más probable del ADN del individuo analizado. En este enfoque, se asume una distribución multinomial y se busca la combinación de poblaciones que mejor explique el genotipo del individuo. Además, utilizando modelos de aprendizaje automático, ajustamos la ascendencia y calculamos los valores de ascendencia de los grupos más específicos. Para más detalles, puede ver el apéndice para Nerds.
Validación
Para investigar la eficacia de la calculadora de ancestralidad, se realizó un muestreo estratificado de cada población, lo que dio como resultado un total de 1.530 individuos de ascendencia conocida que se utilizaron para evaluar las métricas de precisión y sensibilidad del método. La precisión resume qué tan certero es el método a la hora de indicar la ascendencia más alta, mientras que la sensibilidad indica la proporción de la ascendencia más alta indicada correctamente por el método, considerando los individuos de una ascendencia determinada. En otras palabras, la precisión responde a la pregunta:
“Cuando el método asigna la mayor ascendencia a la población X, ¿con qué frecuencia esta asignación refleja realmente la población X?”
La métrica de sensibilidad, por su parte, trata de responder a lo siguiente:
“De las muestras procedentes de la población X, ¿con qué frecuencia el método asigna correctamente la mayor ascendencia de estas muestras a la población X?”
Por ejemplo, en la Tabla 1, vemos que el método tiene una sensibilidad del 99% para los individuos pertenecientes al grupo “América Andina”, es decir, indica correctamente que la mayor proporción de la ascendencia de los individuos es efectivamente de esta región. Por otro lado, para el mismo grupo, encontramos un 93,0% de precisión, revelando que el 7,0% de los casos, analizados como si fueran mayoritariamente de América Andina, tienen otra región de origen como predominante. Esta región de origen suele corresponder a otras poblaciones genéticamente similares, como otros grupos poblacionales nativos americanos, en el caso de la población andina.
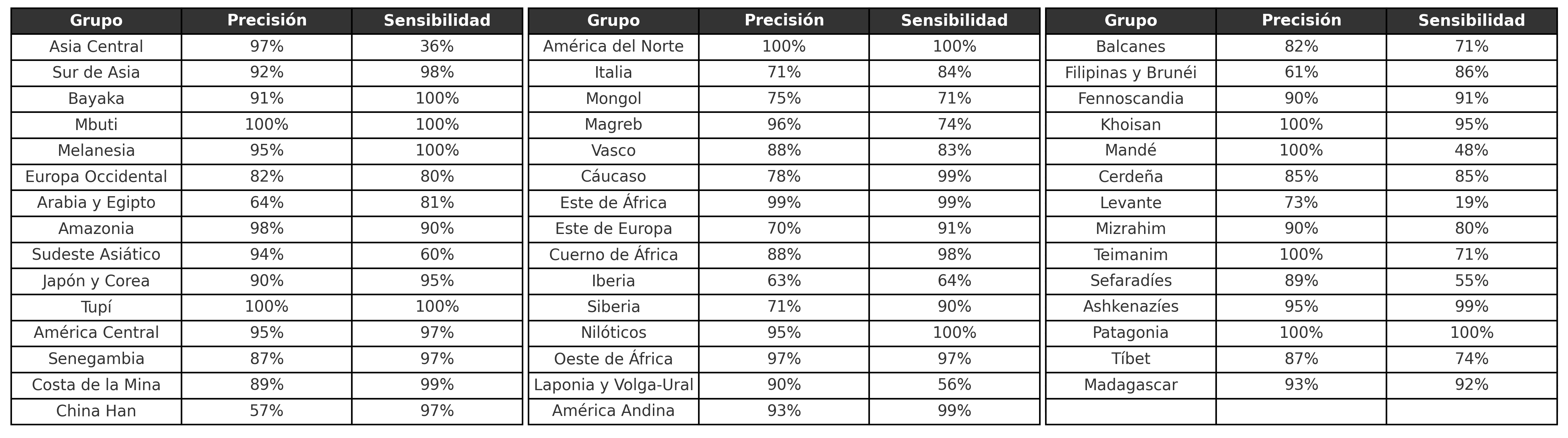
En general, hay un efecto de pérdida-ganancia entre las métricas de precisión y sensibilidad. Por lo general, los sistemas con alta precisión serán muy cautelosos a la hora de indicar la ascendencia más alta. Un posible efecto de esto sería un valor de sensibilidad más bajo, ya que la mayor restricción podría dar lugar a una mayor cantidad de falsos negativos. Nuestra metodología fue capaz de marcar con gran precisión y sensibilidad la ascendencia esperada de los individuos analizados.
Pero ¿cómo debo interpretar mis resultados?
Siempre hay que tener en cuenta que las proporciones obtenidas se refieren al conjunto de poblaciones investigadas. Así, un resultado que indique una proporción más alta para un determinado grupo indica que la mayor parte de su ADN coincide con el perfil observado para ese grupo. Por lo tanto, en algún momento de la historia, algún antepasado de esta región en particular probablemente se unió a su línea familiar.
Es importante señalar que estos valores reflejan que su ADN es similar al de los individuos muestreados para una región determinada. De hecho, es a partir de ellos que se predice el perfil genético de cada grupo utilizado para los cálculos de admixture, es decir, la mezcla genética de las diferentes poblaciones. Por ejemplo, cabe esperar que los individuos del continente americano sean un reflejo de la mezcla de amerindios, europeos, africanos y pueblos de Oriente Medio y diáspora judía, como indica la historia americana de los últimos 500 años.

Parte III
¿Qué pasa con mis datos después?
Tan importante como entender cómo desarrollamos nuestros métodos y llegamos a los resultados de su ancestralidad es entender cómo cuidamos tus datos. En Genera, la seguridad de tus datos es de suma importancia y nuestro objetivo es ser transparentes al respecto. Su información genética se guarda en nuestras bases de datos de forma segura y anónima, de modo que no se permite el acceso externo a la misma. Puede acceder a nuestra política de privacidad, punto por punto, aquí.
Apéndice para Nerds
¿Cómo funciona la estimación de máxima verosimilitud (maximum likelihood estimation – MLE)?

Cuando analizamos el ADN, no sabemos de antemano qué grupos de población han influido en su composición. Para cada SNP analizado, obtenemos como resultado sólo un par de letras, cada una heredada de uno de los padres. Estas letras (A, T, C o G) se denominan alelos. La frecuencia de cada alelo en cada SNP varía de una población a otra, por lo que podemos utilizar esa información para predecir con qué región es más probable que se asocie un determinado fragmento de ADN. Por ejemplo, si el alelo A para un determinado SNP es más común en personas de origen árabe y el individuo analizado tiene el genotipo AA para este SNP, entonces suponemos que debe haber una mayor probabilidad de que tenga ascendencia árabe. Sin embargo, un solo SNP no proporciona suficiente información para distinguir entre grupos de población. Por eso analizamos miles de SNP, relacionando el genotipo del individuo con la frecuencia alélica de cada población. La indicación de la composición ancestral de un individuo se obtiene mediante un algoritmo desarrollado a partir del método de estimación de máxima verosimilitud (MLE). Mediante este método, calculamos el grado de similitud de su ADN con el perfil alélico de 40 regiones geográficas e indicamos la proporción de ascendencia de cada región.
El objetivo del método de máxima verosimilitud es encontrar la distribución de probabilidad que mejor se ajusta a un conjunto de datos observados. Para ello, se ajustan los parámetros de estas distribuciones hasta que empiezan a reflejar el comportamiento observado de los datos. Este ajuste se computa calculando la probabilidad de que los datos pertenezcan a una determinada distribución para valores específicos de los parámetros probados. El conjunto de parámetros más adecuado es, por tanto, el resultado de maximizar el cálculo de estas probabilidades mediante métodos de optimización numérica.
En la figura 1, ilustramos un ejemplo de cómo funciona el método. El conjunto de datos está representado por las posiciones de las bolas azules en la línea horizontal, y las líneas, en color púrpura, son las curvas que representan diferentes intentos de distribuciones de probabilidad (A, B, C y D) para representar los datos. Las líneas de puntos, proyectadas sobre la línea horizontal, representan el valor medio (𝛍) de esa distribución de probabilidad, y la altura de cada parte de la curva representa la densidad de puntos esperada en esa región. En A, se probó una distribución exponencial para explicar el conjunto de datos, sin embargo, se observa que no es fiel al comportamiento de los datos – para que este tipo de distribución sea adecuada, las bolas deberían estar más concentradas en la parte izquierda del eje horizontal, con unas pocas espaciadas hacia la derecha. En B, C y D, se probaron distribuciones normales con diferentes valores de media y desviación estándar (𝞂). Entre ellas, resulta que la distribución D es la que mejor se ajusta a los datos (matemáticamente, la probabilidad de ajuste de los datos a la curva es máxima en D).

Para el cálculo de la composición de la ancestralidad, en concreto, buscamos ajustar los datos genotípicos del individuo a una distribución multinomial. En este caso, el conjunto de parámetros obtenidos con el método refleja la proporción de la contribución de cada grupo de población al individuo analizado.
¿Cómo funciona el ajuste por ascendencia?
El ajuste de la ascendencia se realiza a partir de algoritmos de machine learning. Entrenamos una serie de modelos que se utilizan para afinar la composición de la ancestralidad junto con el método de máxima verosimilitud.
¿Cómo se realiza el cálculo de los grupos más específicos?
El cálculo de la ancestralidad de las subregiones se realiza con modelos de machine learning para cada una de las regiones que se pueden subdividir (Tabla de Poblaciones de Géneros). Este resultado se presenta en base a una escala de 1 a 5, y cuanto más alto sea este valor, mayor será la probabilidad de que la subregión explique la ascendencia encontrada en los pasos anteriores.